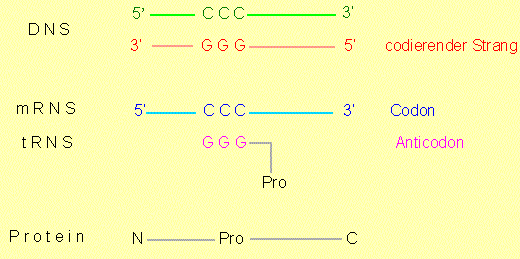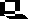
Es besteht ein direkter Zusammenhang zwischen den Nukleotidsequenzen in der Desoxyribonukleinsäure (DNS) und den Aminosäuresequenzen in Proteinen (Polypeptiden). Die Aminosäuresequenz eines Proteins wird demnach durch eine Nukleotidsequenz codiert, oder andersherum ausgedrückt: eine Nukleotidsequenz trägt eine Information, die die Bildung einer Aminosäuresequenz determiniert (instruiert). Die Beziehung zwischen der Abfolge von Nukleotiden und von Aminosäuren wird als genetischer Code bezeichnet.
Nukleotidsequenzen in der DNS sind Abfolgen von vier verschiedenen Nukleotiden (mit den Basen A, T, C, G), Aminosäuresequenzen in Proteinen sind Abfolgen von 20 verschiedenen Aminosäuren. Man kann sich nun die Frage vorlegen: Wie sieht ein Codewort (ein Codon) aus, das eine Aminosäure codiert, wie viele Nukleotide enthält es?
Ein Nukleotid ist offensichtlich zu wenig, denn damit ließen sich nur vier Aminosäuren eindeutig determinieren. Auch Nukleotidpaare (AA, AT, AG...usw.) geben uns nicht genügend Codeworte. Es sind 42 = 16, aber mindestens 20 brauchen wir. Wie sieht es bei einem Triplett aus? AAA, AAT, AAG... 43 = 64 Möglichkeiten stehen rechnerisch zur Verfügung. Das reicht, scheint aber gleichzeitig auch zu viel zu sein. Noch unübersichtlicher wäre die Situation, wenn man Quadrupletts in Erwägung ziehen würde. 44 = 256 Möglichkeiten.
Genetische Experimente und physikalisch-chemische Messungen gaben letztlich den Ausschlag für die Annahme eines Triplettcodes. Damit ist gleichzeitig vorweggenommen, daß alle Codeworte gleich lang sind. Doch wie ist der Code organisiert? Werden alle 64 Codons benötigt? Ist er überlappend, oder nicht. Drei Alternativen wären dabei denkbar:

Die Antwort ließ sich durch eine einfache Überlegung entscheiden. Bei einem überlappenden Code hätte eine Aminosäure in einem Protein einen Einfluß auf die Auswahl der nachfolgenden. Hinter einer, die z.B. durch AAA codiert wird, dürfte bei einem stark überlappenden Code nur eine stehen, die durch AAX codiert würde. Insgesamt gäbe es dafür nur vier Möglichkeiten. Bei einer schwachen Überlappung könnte hinter einer Aminosäure auch wieder nur eine begrenzte Zahl anderer stehen (16 von 20), hinter einer durch AAA codierten, nur solche, die durch AXY codiert würden.
Schon 1957 lagen genügend experimentell ermittelte Aminosäuresequenzen vor, um Nachbarschaftshäufigkeiten auszuwerten. S. BRENNER (Cambridge/England) tat das und kam zu dem Schluß, daß jeglicher überlappende Code ausgeschlossen ist, denn in Proteinen wurde keine Aminosäure durch die in der Sequenz vor ihr stehende beeinflußt.
Ein analoges Beispiel hierzu liefert unsere Sprache. In Worten kann, von einer Ausnahme abgesehen (einem q folgt stets ein u), jeder Buchstabe hinter jedem anderen stehen.
Ein weiteres Problem wäre: Wodurch wird das Startzeichen zum Lesen des Codes gegeben? Schließlich: Welche Ansätze gab es zur Lösung des genetischen Codes?
Einen Code kann man immer nur dann lösen, wenn der Gegner Fehler macht und wenn man merkt, welches System diesen Fehlern zugrunde liegt. Von Fehlern dieser Art ist auch der genetische Code nicht frei. Wir kennen sie als Mutationen. Sie machen sich im simpelsten Fall dadurch bemerkbar, daß in einer Aminosäuresequenz anstelle einer bestimmten Aminosäure eine andere steht. Wenn das Konzept des genetischen Codes richtig ist, müßte demnach in der entsprechenden Nukleotidsequenz ein Nukleotid durch ein anderes ersetzt worden sein.
Man kennt mutationsauslösende Substanzen (Mutagene), die ganz bestimmte, gerichtete Substitutionen von Basen in Nukleinsäuren hervorrufen. Hierzu gehören Nitritionen (Salpetrige Säure), die durch eine Desaminierungsreaktion eine Umwandlung von C nach U oder von A nach G nach sich ziehen.
Für biologische Versuche ist dabei die Menge der Umwandlungen wichtig. Die Einwirkung Salpetriger Säure auf DNS (oder RNS) darf nicht zu lange dauern und ihre Konzentration darf nicht zu hoch sein, denn es darf nur ein geringer Prozentsatz der in den Nukleinsäuren enthaltenen C- und A-Reste verändert werden. Der mutationsauslösende Prozeß ist natürlich ein statistischer Vorgang. Wir wissen zwar, daß C oder A betroffen sind, aber wir wissen niemals im voraus, an welcher Position ein C und/oder A verändert wird. In vielen Fällen wird durch eine solche Modifikation (Basensubstitution) eine lebensnotwendige Information beeinflußt. Es gilt daher die Regel, daß jedes Mutagen stark inaktivierend wirkt und daß man unter den wenigen Überlebenden eine hohe Anzahl von Mutanten erwarten darf.
Spiegeln sich solche durch ein Mutagen induzierte Basensubstitutionen in Veränderungen von Aminosäuren (Aminosäureaustauschen) in Proteinen wider? Dazu brauchen wir ein geeignetes Versuchsobjekt und hierzu bot sich ein Pflanzenvirus, das Tabakmosaikvirus (TMV) an. Man kannte seit 1959 die Aminosäuresequenz seines Hüllproteins. Es besteht aus einer Abfolge von 158 Aminosäuren (Sequenzanalyse: G. SCHRAMM und Mitarbeiter in Tübingen, A. TSUGITA und H. FRAENKEL-CONRAT in Berkeley). H. G. WITTMANN in Tübingen, und A. TSUGITA und H. FRAENKEL-CONRAT in Berkeley, stellten eine große Zahl nitritinduzierter Mutanten her, isolierten einzelne und bestimmten die Aminosäuresequenzen ihrer Hüllproteine. Dabei stellte sich heraus, daß einzelne Aminosäuren im Vergleich zum Ausgangsstamm (dem Wildtyp) verändert waren. Die Ergebnisse ließen sich wie folgt zusammenfassen:
Sie bilden einen weiteren Beleg dafür, daß der Code nicht überlappend ist, denn sonst hätten in den Mutanten nach Veränderung eines Nukleotids zwei (drei) benachbarte Aminosäuren verändert sein müssen. Solche Fälle wurden nie gefunden.
Es ließ sich eine Richtung der Austausche feststellen. Die neu hinzugekommenen Aminosäuren werden durch U- oder G-reichere Codons (Tripletts) codiert als die ursprünglichen.
Die verschiedenen Austausche ließen sich in einer bestimmten Weise anordnen, aus der hervorging, daß es für einzelne Aminosäuren mehrere Codons geben muß. Damit hätten wir eine partielle Antwort auf die Frage, was mit den 64 - 20 = 44 "überflüssigen" Codons geschieht. Auch sie werden benötigt. Man spricht daher von einem "degenerierten Code" und meint damit, daß es für einige Aminosäuren mehrere Codeworte gibt (Degeneration: hier Redundanz).
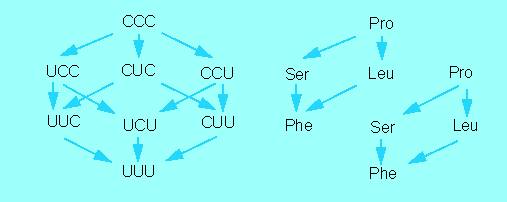
Ansätze zur Entschlüsselung des genetischen Codes. Die Darstellung enthält Austausche, die beim TMV nach Nitritbehandlung erzielt wurden (rechtes Diagramm). Links sind diejenigen Veränderungen in der RNS wiedergegeben, die durch Nitritbehandlung möglich sind, wenn am Ende UUU = Phe herauskommen soll. Weiteres s. Text (Nach H. G. WITTMANN, 1962, 1966)
Wie läßt sich nun aber entscheiden, in welcher Position eines Codons ein C und an welcher ein U einzusetzen ist? Dazu müssen wir einen ganz anderen Ansatz betrachten, der schließlich zur Aufklärung des genetischen Codes führte. Man hatte gelernt, Nukleinsäuren aus freien Nukleotiden zu synthetisieren. A. KORNBERG (Stanford University) isolierte ein Enzym (eine DNS-Polymerase), das an einem DNS-Einzelstrang den komplementären Strang bilden konnte. Der DNS-Einzelstrang dient hierbei als Matrize. S. OCHOA (Rockefeller University, New York) isolierte ein anderes Enzym (eine RNS-Polymerase), das aus Ribonukleotiden RNS synthetisierte, dafür aber keine Matrize benötigte. Die angebotenen Triphosphatnukleotide wurden wahllos zu Polynukleotidketten polymerisiert. Bei einem Angebot von nur UTP oder nur CTP wurden homogene Sequenzen UUUUU... (= PolyU) bzw. CCCC... (= PolyC) gebildet. Bei einem gleichzeitigen Angebot von zwei Nukleotiden (z.B. UTP und CTP) entstanden Polymere, die U und C in einer Zufallsverteilung enthielten.
Was kann man mit solchen synthetischen Polynukleotiden anfangen? Zunächst wenig, doch sehr viel, wenn man über ein System verfügt, mit dem die in ihnen gespeicherte Information gelesen werden kann. M. NIRENBERG und H. MATTHEI (1961, am National Institute of Health, Bethesda) entwickelten ein zellfreies (in vitro) System, das zu einer Proteinbiosynthese befähigt war. Dazu werden benötigt: RNS, Ribosomen, ein löslicher Überstand aus einem Bakterienextrakt (von Escherichia coli), Aminosäuren sowie ATP, CTP, GTP u.a. Wesentlich sind im Augenblick zwei Aspekte:
Unter in vitro-Bedingungen war eine Proteinsynthese nachweisbar. Der Test hierzu war zunächst sehr einfach: Man setzte einzelne radioaktiv markierte Aminosäuren zu und prüfte, ob sich die Radioaktivität nach einer kurzen Inkubationszeit durch Trichloressigsäure (TCA) ausfällen ließ. Man weiß, daß freie Aminosäuren durch TCA-Zugabe nicht fällbar sind, während Proteine ausfallen.
Durch gezielte Zugabe einer bestimmten genetischen Information, z.B. durch PolyU (UUUUU...), ließ sich ausschließlich die Aminosäure Phe in eine TCA-fällbare Form überführen. Damit war das erste Codewort enträtselt: UUU = Phe.
Wir können jetzt zu den Befunden an Mutanten des Tabakmosaikvirus zurückkehren und erkennen, daß Leu und Ser durch C-reichere Codons (UUU, UCU oder CUU) codiert werden und daß der C-Gehalt der Codons von Pro, Ser und Leu noch höher sein muß (CCU, CUC, UCC oder CCC). Durch Vergleich mit weiteren Ergebnissen, die im zellfreien System ermittelt wurden, konnte auch die genaue Reihenfolge der Nukleotidbasen in jedem Codon festgelegt werden.
Eine abschließende Antwort auf noch ausstehende Fragen und die vollständige Aufklärung des genetischen Codes gelang, nachdem man in dem eben skizzierten zellfreien System genau definierte, in ihrer Basenzusammensetzung und -sequenz determinierte (synthetische) Polynukleotide testen konnte. 1963 wurden die Arbeiten erfolgreich abgeschlossen.
Die Tabelle gibt die Zuordnung aller 64 Codons zu den entsprechenden Aminosäuren wieder. Die Ziffern 1, 2 und 3 beziehen sich auf die Position des Nukleotids im Codon. z.B. 1 = A, 2 = C, 3 = A: ACA = Thr. Drei der Codons "amber", "ochre" und "opal" stellen Signale für Kettenabbruch dar. Ein weiteres, AUG, das normalerweise für Met codiert, kann auch Kettenanfang bedeuten. In der linken oberen Ecke stehen hydrophobe Aminosäuren, in der rechten unteren hydrophile.
| 2 | |||||||||
| U | C | A | G | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | U | Phe | Ser | Tyr | Cys | U | 3 | ||
| Phe | Ser | Tyr | Cys | C | |||||
| Leu | Ser | ochre | opal | A | |||||
| Leu | Ser | amber | Trp | G | |||||
| C | Leu | Pro | His | Arg | U | ||||
| Leu | Pro | His | Arg | C | |||||
| Leu | Pro | Gln | Arg | A | |||||
| Leu | Pro | Gln | Arg | G | |||||
| A | Ile | Thr | Asn | Ser | U | ||||
| Ile | Thr | Asn | Ser | C | |||||
| Ile | Thr | Lys | Arg | A | |||||
| Met* | Thr | Lys | Arg | G | |||||
| G | Val | Ala | Asp | Gly | U | ||||
| Val | Ala | Asp | Gly | C | |||||
| Val | Ala | Glu | Gly | A | |||||
| Val | Ala | Glu | Gly | G | |||||
Aus den Ergebnissen lassen sich eine Anzahl von Schlußfolgerungen ziehen:
Alle 64 Codons werden genutzt. 61 können bestimmten Aminosäuren zugeordnet werden, drei dienen als Stoppsignal, eines (AUG) alternativ als Aminosäurecodon oder Startsignal.
Die Zahl der Codons für die einzelnen Aminosäuren ist unterschiedlich, für einige, wie Met und Trp, gibt es nur ein, für viele zwei oder vier, und für einige (Ser, Arg) sogar sechs Codons. Es besteht eine Korrelation zwischen Häufigkeit von Codons und Häufigkeit der entsprechenden Aminosäuren in Proteinen. Eine Ausnahme bildet dabei lediglich die Aminosäure Arg, für die es sechs Codeworte gibt, die aber in bezug dazu in Proteinen unterrepräsentiert ist.
Die Codons sind den Aminosäuren nicht wahllos zugeordnet. Die beiden ersten Nukleotide eines Codons haben einen höheren Informationswert als das dritte, z.B. stehen GUU, GUC, GUA und GUG alle für Val. UC-reiche Codons (Tripletts) codieren für hydrophobe, AG-reiche für hydrophile Aminosäuren. Der genetische Code ist demnach als extrem konservativ einzustufen.
Viele (nahezu 30%) der Basensubstitutionen ändern nichts an den Codierungseigenschaften, z.B.
UUU > UUC: Phe > Phe
Selbst wenn eine Basensubstitution einen Aminosäureaustausch hervorruft bleibt der chemische Charakter des Seitenkettenrests in den meisten Fällen gewahrt (konservative Austausche):
UUU > UUG: Phe > Leu
CUC > AUC: Leu > Ile
AAA > AGA: Lys+ > Arg+
AAA > GAA: Lys+ > Asp-
Natürlich gibt es Ausnahmen (radikale Austausche) wie z.B.:
GAG > GUG: Glu- > Val
GAA > GUA: Glu- > Val
Die letzte Kategorie führt in der Regel zu funktionslosen oder nur mangelhaft funktionierenden Proteinen. Da sie der Selektion unterworfen sind, haben derartige Mutanten unter natürlichen Bedingungen keine oder nur eine reduzierte Überlebenschance.
Im Verlauf der Evolution hat sich also ein genetischer Code herausgebildet, der Stabilität gewährt und der so gestaltet ist, daß eine Anzahl von Änderungen im Protein gar nicht in Erscheinung tritt. Auch die Zahl der Codons für einzelne Aminosäuren ist nicht dem Zufall überlassen. Es wurde zwar schon gesagt, daß häufig in Proteinen auftretende Aminosäuren durch mehr Codons repräsentiert sind als die selteneren. Die Frage ist nur, was ist Ursache und was ist Wirkung? Ohne eine klare Antwort darauf geben zu können, läßt sich zumindest eine weitere Korrelation anführen: Am wenigsten Codons gibt es für die Aminosäuren, deren Biosynthese aufwendiger ist als die der anderen, d.h., daß für deren Synthese mehr Energie investiert werden muß als für die einfacheren (und damit häufigeren). Als Ausnahme bleibt auch hier wieder das Arginin zu nennen.
Die Befunde am Tabakmosaikvirus, im Vergleich zu denen, die an Mikroorganismen und später an eukaryotischen Zellen ermittelt wurden, machen deutlich, daß der genetische Code universell ist, d.h., die in der Tabelle aufgelistete Zuordnung von Codons und Aminosäuren ist für alle Organismen (Mikroorganismen, Tiere, Pflanzen) gleich. Eine Ausnahme wurde schließlich doch noch gefunden: In tierischen Mitochondrien gespeicherte Information wird aufgrund eines andersartigen Lesemechanismus in einigen Fällen anders genutzt:
AUU: statt Ile: Met
AUA: statt Ile: Met
UGA: statt stop: Trp
AGA: statt Arg: stop
AGG: statt Arg: stop
Auch in pflanzlichen Mitochondrien wurden Abweichungen festgestellt. Offenbar gibt es dort sogar artspezifische Unterschiede. Bei Oenothera steht UGA (als TGA in der DNS identifiziert) für Termination und CGG für Trp (W. SCHUSTER, A. BRENNICKE, 1985).
Weitere Einzelheiten hierzu:
Wir müssen uns jetzt natürlich fragen, wie die genetische Information übersetzt wird, wie also der Informationsfluß aussieht? Normalerweise handelt es sich dabei um einen Zweistufenprozeß:
Die in der DNS gespeicherte Information wird in RNS überschrieben (=Transkription). Die Überschreibung erfolgt nicht in einem Stück; es werden jeweils nur Teilinformationen bearbeitet.
Die nunmehr in RNS enthaltene (Teil)-Information wird in einem komplexen Vorgang, an dem eine Vielzahl von Komponenten mitwirken, in Protein übersetzt (= Translation, Proteinbiosynthese).
In den letzten 2 Jahrzehnten sind eine Reihe effizienter Methoden zur Sequenzierung von Genen und vollständigen Genomen entwickelt worden. Die Nukleotidsequenzen werden in Datenbanken gespeichert (daily update) und sind dort jedcerzeit frei zugänglich. Es gibt eine Anzahl von Programmen, mit denen die Daten bearbeitet und untereinander verglichen werden können. Sequenzvergleiche sind möglich, ebenso wie die Zuordnung zu Genen bzw. potentiellen Genen: open reading frames (ORF) mit Startcodon beginnend und mit Stoppcodon schließend.
Die zentrale Datenbank in Europa ist: EMBL Outstation: European Bioinformatics Institute in Hinxton bei Cambridge / England. Nukleotid- und Aminosäuresequenzen sind am besten über das Sequence Retrival System erreichbar. Das EBI dient auch als Mirror Site für weitere große internationale Datenbanken der Genetik und Molekularbiologie. Erreichbar unter:
 Die zentralen Datenbanken in den USA sind GeneBank und in Japan DDBJ (DNA Data Bank of Japan).
Eine gute Zusammenstellung der Daten bietet die Kyoto Encyclopedia of Genes and Genomes (KEGG) auf deren Datenbestände an verschiedenen Stellen in Botanik online zurückgegriffen wird.
So werden an dieser Stelle als Beispiele die vollständigen Genome des Bakteriums Escherichia coli und der Bäckerhefe Saccharomyces cerevisiae präsentiert.
Die zentralen Datenbanken in den USA sind GeneBank und in Japan DDBJ (DNA Data Bank of Japan).
Eine gute Zusammenstellung der Daten bietet die Kyoto Encyclopedia of Genes and Genomes (KEGG) auf deren Datenbestände an verschiedenen Stellen in Botanik online zurückgegriffen wird.
So werden an dieser Stelle als Beispiele die vollständigen Genome des Bakteriums Escherichia coli und der Bäckerhefe Saccharomyces cerevisiae präsentiert.
Vorsicht bei der Nutzung der nachfolgenden Daten:. Es handelt sich in allen Fällen um komplexe Java-Skripts (20 MB Arbeitsspeicher erforderlich) mit Links auf die Original URL in Kyoto. Das vollständige Escherichia coli- Genom
und die Aminosäuresequenzen der codierten Proteine sind zusätzlich im vollen Umfang in einer einzigen Datei widergegeben (Größe: 10,9 MB). Einzelheiten zur Nutzung sind im Manual genannt. Die Genbenennungen sind den Genomkatalogen zu entnehmen.
|
|